

Pituitary adenomas
What is Pituitary Adenomas?
Pituitary tumors are abnormal growths that develop in your pituitary gland. Some pituitary tumors result in too much of the hormones that regulate important functions of your body. Some pituitary tumors can cause your pituitary gland to produce lower levels of hormones.
Most pituitary tumors are noncancerous (benign) growths (adenomas). Adenomas remain in your pituitary gland or surrounding tissues and don't spread to other parts of your body.
There are various options for treating pituitary tumors, including removing the tumor, controlling its growth and managing your hormone levels with medications. Your doctor may recommend observation — or a ''wait and see'' approach.
What are Pituitary Adenomas
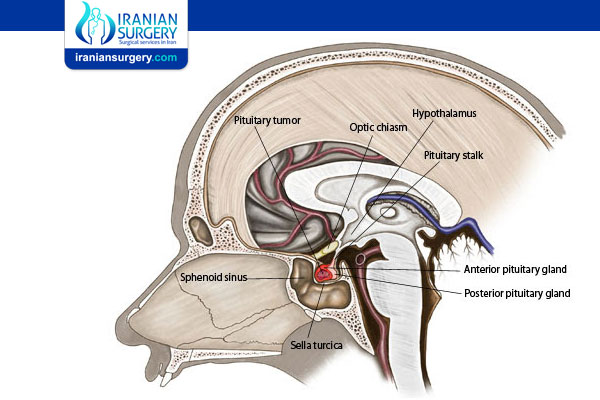
What is the pituitary?
The pituitary is a gland about the size of a pea that is joined to the base of the brain. It is found behind the nose and the sphenoid sinus (air space behind the face), right below another important and related structure called the hypothalamus.
A gland is an organ that makes and releases special substances, such as hormones, that act on other organs and tissues to make them work. The pituitary is called the "master gland" because its hormones regulate the balance of hormones made by most of the other glands in the body. This way, the pituitary controls many processes, such as growth, development, and reproduction. It also controls the function of certain organs, such as the kidneys, breasts, and uterus.
The pituitary has three parts or lobes, each with its own job in the body. The front lobe, the anterior pituitary, makes up about 80% of the gland and is found closest to the front of the head. Its job is to make and release many "signaling" hormones into the bloodstream. These hormones move through the blood to other glands and organs, where they signal the organs and glands to start or stop working.
The intermediary lobe secretes only one hormone. This hormone affects skin pigmentation. The back part of the gland, the posterior pituitary, does not make any hormones itself. Instead, it contains nerve endings of brain cells that come from the hypothalamus. These brain cells make the hormones, which then move down to and are stored in the posterior pituitary for later use.
The hypothalamus and the pituitary work hand-in-hand. The hypothalamus makes hormones that, in turn, signal the pituitary to release its hormones.
What Is a Pituitary Adenoma?
A pituitary adenoma is a growth or tumor on the pituitary. Most pituitary adenomas are slow-growing and benign, which means they are not cancer and do not spread to other parts of the body. However, as they grow big they can put pressure on nearby structures, such as the nerves that connect the eyes to the brain, and cause symptoms. This is known as the “mass effect.”
A large adenoma can also crush normal pituitary cells and keep them from working properly, leading to a condition called hypopituitarism. This condition can cause low blood pressure, tiredness, and changes in your sex drive and function. It can also make you feel less able to manage stress.
Pituitary adenomas are labeled based on their size. Tumors smaller than 1 cm are called microadenomas. Tumors bigger than 1 cm are called macroadenomas. Many adenomas cause symptoms by releasing extra hormones into the bloodstream, but not all adenomas make hormones.
Some, called non-functioning or null cell adenomas, do not make hormones. About one-third of all pituitary tumors are non-functioning adenomas. They usually become large and are found because they cause symptoms due to the mass effect.
How Common Are Pituitary Adenomas?
Pituitary adenomas make up 10% to 15% of all tumors that develop within the skull. They are found in about 77 out of 100,000 people, although it is believed that they actually occur in as many as 20% of people at some point in their lives. However, many pituitary adenomas, especially microadenomas, do not cause serious symptoms and are never found. Macroadenomas are about twice as common as microadenomas. Pituitary adenomas can occur at any age, but are more common in people in their 30s or 40s. They are rarely found in those younger than 20. Women get adenomas more often than men.
Symptoms And Causes of Pituitary Adenomas
What Causes Pituitary Adenomas?
The exact cause of pituitary adenomas is not known. However, some have been linked to accidental changes in DNA, the material within a cell that makes up our genes. (Genes are the directions for making the proteins that control a cell's function.) These changes cause the cells in the pituitary to be abnormal and grow out of control, making a tumor. The changes can be passed down from parents to their children (heredity), but usually happen on their own sometime during a person's life.
What Are the Symptoms of A Pituitary Adenoma?
The symptoms of pituitary adenomas vary depending on the type of hormone released by the tumor or its mass effect on nearby structures. Having too many hormones can lead to certain disorders or syndromes, including:
. Gigantism in children and acromegaly in adults from too much growth hormone (GH)
. Cushing’s disease from too much adrenocorticotropic hormone (ACTH), which triggers the adrenal glands to make steroid hormones
. Hyperthyroidism from too much thyroid stimulating hormone (TSH)
Some general symptoms of pituitary adenomas include:
. Headaches
. Vision problems (double vision, vision loss)
. Nausea or vomiting
. Changes in behavior, including hostility, depression and anxiety
. Changes in the sense of smell
. Nasal drainage
. Sexual dysfunction
. Fatigue (extreme tiredness)
. Unexplained weight gain or loss
. Achy joints or muscle weakness
. Early menopause
. Changes in your monthly periods (women)
How Are Pituitary Adenomas Diagnosed?
If your doctor thinks you might have a pituitary adenoma, he or she will do a full review of your symptoms and your medical background, and will perform a physical exam. Lab tests might be done to check the hormone levels in your blood. An imaging test, such as an MRI or CT scan*, can show a growth on the pituitary. These tests can confirm the diagnosis of a pituitary adenoma. If you are having problems with your sight, the doctor might also have you take a visual field test to check your eye function. Sometimes, pituitary adenomas are found by accident when an MRI or CT is being done for another condition.
*An MRI uses a large magnet, radio waves, and a computer to create clear images of the body. CT uses computers to combine many X-ray images into cross-sectional views of the inside of the body.
How Are Pituitary Adenomas Treated?
Pituitary adenomas are usually treated with surgery, medicine, radiation or a combination of these therapies.
. Surgery: Doctors can often remove the tumor with endoscopic surgery, reaching the pituitary using very small tools and a camera inserted in a small opening made through the nose and sinuses. This approach is called the transsphenoidal technique. In cases where this approach cannot be used, the doctor must open the skull to get to the pituitary and the adenoma.
. Medical management: With some types of pituitary adenomas, the tumor can be treated with medication that shrinks the tumor and relieves symptoms.
. Radiation: Radiation therapy uses high-energy X-rays to kill cancer cells and shrink tumors. A special form of radiation therapy, called stereotactic radiosurgery, uses a high dose of radiation aimed precisely at the adenoma from more than one direction to keep the tumor from growing.
In some cases, the doctor may choose to use yearly tests to check that the adenoma is not growing, while watching the patient for any symptoms. This might happen with small tumors that are not causing serious symptoms and with people who have other major medical conditions.
Can Pituitary Adenomas Be Prevented?
Pituitary adenomas cannot be prevented.
What Is the Outlook for People with Pituitary Adenomas?
The outlook depends on the size and type of adenoma you have. When treatment destroys the tumor, most patients who have benign adenomas can return to full, normal lives. Adenomas can recur, which means you will need treatment again. About 18% of patients with non-functioning adenomas and 25% of those with prolactinomas, the most common type of hormone-releasing adenomas, will need more treatment at some point. In some cases, adenoma treatment results in low hormone levels, and you have to take hormone medicines to replace what you have lost.
When Should I Call My Healthcare Provider?
Call your healthcare provided if you have problems with your vision, have headaches that don't go away or keep coming back, or have any of the other symptoms of a pituitary adenoma.
Source:
https://my.clevelandclinic.org/health/diseases/15328-pituitary-adenomas-